
آنیوپلوئیدی کروموزومی چیست؟ (Chromosomal Aneuploidy)
تقریبا تمام سلول های بدن انسان دارای 23 جفت کروموزوم (46 کروموزوم) هستند که نیمی از آن از پدر و نیم دیگر از مادر دریافت شده است. تمام اطلاعاتی که بدن انسان برای رشد و تکوین نیاز دارد درون کروموزوم ها وجود دارد. هر کروموزوم حاوی هزاران ژن است که اطلاعات مربوط به ساخت پروتئین های مورد نیاز برای رشد، تکوین و واکنش های بیوشیمیایی را ذخیره می کند. بنابراین هرگونه اختلال و ناهنجاری حتی در یک کروموزوم انسان (مانند آنیوپلوئیدی ها) موجب مختل شدن تعادل سلول ها و در نتیجه ایجاد اختلال در زندگی طبیعی فرد می شود. سلول ها به مکانیسم های دقیق برای اطمینان از جداسازی دقیق کروموزوم ها در طول میتوز و میوز برای حفظ حالت euploidy خود متکی هستند و بروز هرگونه اشتباه در این روند منجر به بروز انواع مختلفی از ناهنجاری های کروموزومی می گردد.
ناهنجاری های کروموزومی می توانند به صورت تصادفی هنگام تشکیل تخمک یا اسپرم یا در مراحل اولیه رشد جنین رخ دهند، البته سن مادر و برخی عوامل محیطی دیگر نیز ممکن است در بروز خطاهای ژنتیکی نقش داشته باشد. آنیوپلوئیدی ناشی از تفکیک نادرست کروموزوم ها در طول میوز یکی از دلایل اصلی ناباروری و نقایص مادرزادی ارثی است. از سوی دیگر، حدود یک سوم از موارد سقط جنین در زنان به دلیل وجود این دسته از ناهنجاری ها در ژنوم افراد می باشد. به طور کلی، آنیوپلوئیدی ها به دو گروه ناهنجاری های ساختاری و تعدادی کروموزوم ها طبقه بندی می شوند که عبارتند از:
-
ناهنجاری های ساختاری (Structural abnormalities)
به دسته ای از ناهنجاری ها گفته می شود که در آن بخشی از کروموزوم دچار جهش های مختلفی از جمله حذف (deletion)، مضاعف شدگی (duplication)، درج (insertion)، وارونگی (inversion) و یا جهش های نقطه ای (Point mutation) می گردد.
-
ناهنجاری های عددی (Numerical abnormality)
در این دسته از ناهنجاری های کروموزومی تعداد طبیعی کروموزوم ها دچار تغییر شده است، به عنوان مثال به جای وجود 2 عدد از یک کروموزوم فرد دارای یک (مونوزومی) یا سه عدد (تریزومی) از آن می باشد. این حالت شایع ترین نوع ناهنجاری های کروموزومی است که به آنها آنیوپلوئیدی گفته می شود و سندرم داون یا تریزومی 21 احتمالاً شناخته شده ترین مثال آنیوپلوئیدی کروموزومی می باشد.
علت بروز ناهنجاری های تعدادی کروموزم ها
در زمان تقسیم سلولی باید کروموزوم ها به دقت به سلول های دختری منتقل شوند تا از بروز آنیوپلوئیدی ها جلوگیری شود. بروز آنیوپلوئیدی ها می تواند علل مختلفی داشته باشد که برخی از آنها عبارتند از:
-
خطا در اتصال میکروتوبول ها به کینه توکور
تفکیک کروموزوم ها (chromosome segregation) از طریق اتصال دقیق رشته های دوک تقسیم و میکروتوبول ها از سانتریول های دو سمت سلول به ناحیه ای به نام کینه توکور (ساختاری پروتئینی در ناحیه سانترومر کروموزوم ها) در کروموزوم ها انجام می شود. برای جداسازی دقیق کروموزوم ها باید کینه توکورهای کروماتیدهای خواهری از دو سمت سانترومر به میکروتوبول ها متصل شده و در مرحله آنافاز به قطبین سلول حرکت کنند. در افراد مبتلا به تریزومی اتصالات نادرست کینه توکور-میکروتوبول (kinetochore–microtubule attachment) یا K-MT منجر به حرکت هردو کروماتید خواهری به یک سلول دختری می گردد.
-
نقص در کوهسین (Cohesion)
در طول همانند سازی DNA کروماتیدهای خواهری یک کروموزوم از طریق نوعی پروتئین چند زیر واحدی به نام کوهسین متصل به هم باقی می مانند. در زمان تقسیم سلولی و در مرحله آنافاز، برش پروتئولایتیک (Proteolytic cleavage) باعث از بین رفتن پروتئین کوهسین شده و امکان جداسازی کروماتیدهای خواهری فراهم می شود. نتایج مطالعات مختلف در مخمر نشان داده شده است که کروماتیدهای خواهری تنها زمانی می توانند به طور مناسبی کنار هم قرار گیرند که پروتئین کوهسین میتوزی SCC1 قبل از فاز S بیان شود. سپس در مرحله آنافاز آنزیم Protease separase با تخریب SCC1 منجر به جداسازی کروماتیدهای خواهری می گردد. باید توجه داشت که آنزیم Protease separase نمی تواند نوع جهش یافته SCC1 را تخریب کند و کروماتیدهای خواهری از یکدیگر جدا نخواهند شد، بنابراین بروز جهش در ژن کد کننده کوهسین می تواند عاملی برای بروز ناهنجاری های تعدادی کروموزوم ها باشد.
-
وجود سانتروزوم های اضافی (Supernumerary centrosomes)
سانتروزوم ها، اندامک های درون سلولی هستند که مسئول تشکیل و سازمان دهی میکروتوبول ها در زمان تقسیم سلولی می باشد. هر سانتروزوم متشکل از دو سانتریول است که به طور عمود بر یکدیگر با زاویه 90 درجه درون سلول قرار دارند. سانتروزوم ها در حین چرخه سلولی و در فاز S تکثیر شده و در زمان تقسیم سلولی به قطبین سلول حرکت می کنند تا پس از اتمام چرخه سلولی، هر سلول دختر دارای یک سانتروزوم باشد و کروماتیدهای خواهری به درستی از یکدیگر جدا شوند. نقص در چرخه تکثیر سانتروزوم که باعث تکثیر بیش از حد این اندامک و افزایش احتمال تشکیل دوک های چندقطبی (multipolar spindles) می گردد، در نتیجه جداسازی کروموزوم ها دچار اختلال شده و می تواند منجر به آنیوپلوئیدی شود.
-
تتراپلوئیدی
تتراپلوئیدی نوعی ناهنجاری تعدادی کروموزوم هاست که فرد به جای داشتن دو سری از کروموزوم ها (2n)، دارای 4 سری (4n) می باشد. این دسته از سلول ها مشتق شده از سلول های دیپلوئید بوده و دارای سانترومر اضافه می باشند که می تواند منجر به بوز آنیوپلوئیدی گردد. یکی از عواملی که منجر به بروز تتراپلوئیدی شدن سلول های می شود، وجود نقص در مرحله سیتوکینز است. از دیگر عوامل دخیل می توان به همجوشی سلولی (cell fusion) غیربرنامه ریزی شده و endoreplication (همانند سازی DNA سلولی بدون انجام تقسیم میتوز) اشاره کرد که منجر به تتراپلوئیدی شدن و بروز آنیوپلوئیدی در تقسیمات بعدی سلولی می گردد.
-
بروز اختلال در عملکرد تلومر
تلومرها ساختارهای تخصصی در انتهای کروموزوم های خطی هستند که اختلال در عملکرد آنها با ایجاد آنیوپلوئیدی مرتبط است. تلومر توسط آنزیمی به نام تلومراز (Telomerase) که نوعی رونوشت بردار معکوس (Reverse Transcriptase) است به انتهای کروموزوم ها اضافه می شود، در صورتی که این آنزیم دچار اختلال گردد کروموزوم های بدون تلومر می توانند از دو انتها به یکدیگر متصل شده و یک کروموزوم دو سانترومری (dicentric chromosomes) ایجاد گردد. کروموزوم دو سانترومری (dicentric chromosomes)، کروموزوم های ناهنجاری هستند که احتمال تفکیک به یک سلول دختر و یا شکسته شدن آنها در بین دو سلول دختر بسیار زیاد است.
علت بروز ناهنجاری های ساختاری کروموزم ها
ناهنجاری های ساختاری اغلب به دلیل بروز جهش های مختلف مانند جابه جایی نامتعادل (non-balanced translocations)، حذف (Deletion)، مضاعف شدن (duplication) و یا شکستگی ناحیه ای از DNA رخ می دهد. به طور کلی آنیوپلوئیدی ها ساختاری با مکانیسم های مختلفی رخ می دهند که یکی از آنها ردیف شدن نادرست کروموزوم های همولوگ (misalignment of homologous chromosomes) در زمان تقسیم میوز و کراسینگ اور نابرابر بین کروماتیدهای غیر خواهری می باشد که منجر به بروز مضاعف شدن در یک ناحیه از کروموزوم می گردد. از دیگر علل بروز ناهنجاری های ساختاری کروموزوم های می توان به نوترکیبی همولوگ غیر آللی نامناسب (Improper non-allelic homologous recombination) اشاره کرد که باعث ایجاد تغییرات در تعداد کپی DNA به دلیل حذف یا تکرار می گردد.
شایع ترین آنیوپلوئیدی ها در انسان
شایع ترین آنیوپلوئیدی ها در انسان، تریزومی یا سه تایی شدن کروموزوم است که باعث می شود تعداد کل کروموزوم های فرد به 47 برسد. تریزومی ها اغلب کشنده بوده و عامل حدود 35 درصد سقط های خود به خودی هستند. تریزومی کروموزوم های 13 (عامل سندروم پاتو) و 18 (عامل سندروم ادوارد) نیز منجر به مرگ کودک در ماه های اولیه زندگی می شوند. ناهنجاری های تعدادی که با وجود آن ها ادامه بارداری ممکن بوده و امکان تولد نوزاد زنده وجود دارد عبارتند از تریزومی 21 (سندروم داون)، مونوزومی X (سندروم ترنر)، سندروم کلاین فلتر (XXY)، سندروم جاکوب (XYY) و تریزومی X (XXX) که در ادامه به توضیح آنها پرداخته می شود.
-
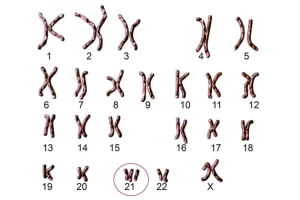
تریزومی 21 یا سندرم داون (Down syndrome)
سندرم داون یکی از اصلی ترین علل بروز ناتوانی های ذهنی در افراد است که نرخ شیوع آن در جمعیت های مختلف متفاوت بوده و حدود 1 در 319 تا 1 در 1000 نوزاد متولد شده می باشد. خطر بروز سندرم داون با افزایش سن مادر بالا می رود و حدود 50 تا 75 درصد از جنین های مبتلا به این بیماری پیش از تولد سقط می شوند. این اختلال ژنتیکی شایع ترین آنیوپلوئیدی در افراد زنده است که می تواند به دلیل وجود تمام یا بخشی از کروموزوم 21 اضافی در فرد ایجاد شود.
علائم
- کوتاه بودن گردن
- چشم های بادامی شکل متمایل به بالا
- یک خط در کف دست
- کوچک بودن دست ها و پاها
- کوتاهی قد
- داشتن تون عضلاتی (muscle tune) ضعیف و یا شل شدن مفاصل
- گوش های کوچک
- زبان بیرون زده
- پلک های چشمی مایل به سمت بالا
- لکه های سفید ریز روی قسمت رنگی چشم به نام Brushfield’s spots

انواع سندرم داون
- تریزومی 21: شایع ترین نوع این بیماری است که حدود 95% از بیماران را شامل شده و این افراد دارای 3 عدد از کروموزوم 21 در تمام سلول های خود هستند.
- ترانسلوکاسیون (Translocation) سندرم داون: حدود 3% از بیماران مبتلا به این نوع از سندرم داون هستند که در آنها تنها بخشی از کروموزوم 21 به کروموزوم دیگر جابه جا شده و ترانسلوکاسیون رخ داده است.
- موزائیک سندرم داون (Mosaic Down syndrome): این حالت در تنها 2% از بیماران دیده می شود که برخی از سلول های فرد دارای 3 عدد از کروموزوم 21 و برخی دیگر 2 عدد از این کروموزوم هستند.
علت
- ایزوکروموزومی 21
- جابه جایی رابرتسونین (2 تا 4 درصد موارد) بین کروموزوم های 21 و 14
- تریزومی 21 و افزایش سطح بیان ژن (increase gene dosage) Hsa21
-
سندروم ادوارد (Edwards Syndrome)
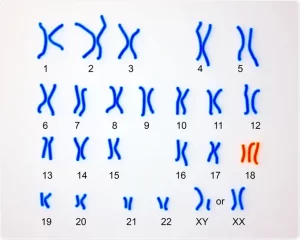
سندروم ادوارد یا تریزومی 18 شایع ترین تریزومی پس از سندروم داون می باشد. این سندروم به دلیل وجود یک کروموزوم 18 اضافی در سلول های بدن ایجاد می شود و کاریوتایپ افراد مبتلا به صورت (47,XY+18 یا 47,XX+18) می باشد. لین بیماری به دلیل خطا در تقسیم سلولی هنگام تشکیل تخمک یا اسپرم ایجاد می شود و در آن وجود یک کروموزوم 18 اضافی الگوی طبیعی رشد سلول را مختل می کند. نرخ شیوع آن در کشور های مختلف متفاوت می باشد و حدود 1 در 3600 تا 10000 تولد زنده است که احتمال وقوع آن با افزایش سن مادر افزایش می یابد. تعداد قابل توجهی از جنین های مبتلا به این اختلال هرگز متولد نمی شوند. تنها 50% نوزادان مبتلا زنده می مانند که پس از تولد در بخش مراقبت های ویژه نگهداری می شوند. بسیاری از نوزادان زنده مانده نیز در روزها و هفته های ابتدایی زندگی می میرند. شانس زنده ماندن دختران بیشتر از پسران بوده و به طور کلی سندروم ادوارد در دختران شایع تر است.
علائم
- مشکلات قلبی مادرزادی
- هیپوتونی
- مشکلات تغذیه ای
- تاخیر در رشد داخل رحمی
- ظاهری لاغر همراه با هیپوتروفی
- میکروسفالی با جمجمه باریک و دولیکوسفالی (dolichocephaly)
- میکرورتروگناتیا (microretrognathia)
- هایپرتلوریسم (hypertelorism)
- گوشهای زاویهدار (angular ears)
- مشاهده برخی مشکلات چشمی مانند میکروفتالمیا ((microphthalmia و کلوبوما (Coloboma)

انواع سندرم ادوارد و علل بروز
- تریزومی کامل کروموزوم 18 به دلیل خطا در میوز II که اغلب مرتبط با افزایش سن مادر می باشد. این حالت در 95% موارد مشاهده می شود.
- تریزومی 18 موزائیک دومین حالت شایع این بیماری است که در کمتر از 5% از بیماران مشاهده می شود.
- تریزومی 18 جزئی (partial trisomy 18) در حدود 2% از مبتلایان دیده شده و تنها از بخشی از کروموزوم 18 سه نسخه در فرد مشاهده می شود.
-
سندروم پاتو (Patau Syndrome)
سندروم پاتو یا تریزومی 13 به دلیل حضور یک کروموزوم 13 اضافی ایجاد می شود و آرایش کاریوتایپ افراد مبتلا به آن به صورت 47,XY+13 یا 47,XX+13 است. این بیماری نسبت به سندروم های داون و ادوارد بسیار نادرتر است و احتمال وقوع آن 1 در هر 16000 تولد زنده است. احتمال وقوع آن با افزایش سن مادر بالا می رود و کروموزوم 13 اضافی منشا مادری دارد. سندروم پاتو نیز مانند سایر آنیوپلوئیدی ها به دلیل خطا در تقسیم سلولی تخمک یا اسپرم و یا بعد از لقاح در سلول تخم ایجاد می شود.
علائم
- نقص در قسمت آهیانه ای-پس سری پوست سر
- هولوپروزنسفالی یا نقص صورت مانند نزدیکی چشم ها، کوچک بودن سر و شکاف کام و لب
- انگشتان اضافه در دست و پا (polydactyly)
- انگشتان خمیده (مشت بسته)
- اختلالات کلیوی
- نابینایی
- ناشنوایی
- همچنین ناهنجاری های دستگاه تناسلی مانند عدم نزول بیضه ها و ناهنجاری کیسه بیضه در مردان و ناهنجاری در شکل رحم در زنان نیز در افراد مبتلا
- ابتلا به بیماری های قلبی مانند نقص دیواره بین بطنی (ventricular septal defect)، نقص تیغه بین دهلیزی (atrial septal defect)، tetralogy of Fallot ، نقص تیغه بین دهلیزی (atrioventricular septal defect) و بطن راست دو خروجی (double outlet right ventricle)

علت بروز
- عدم تکیک کروموزوم ها در میوز (nondisjunction in meiosis)
- جابجایی نامتعادل رابرتسونین (unbalanced Robertsonian translocation) است که منجر به دو نسخه طبیعی از کروموزوم 13 و یک بازوی طولانی دیگر از کروموزوم 13 می شود
- یکی دیگر از علل کمتر شایع موزائیسم است که منجر به 3 کپی از کروموزوم 13 در برخی سلول ها و دو کپی در سایر سلول ها می شود.موزائیسم نتیجه یک خطای عدم تفکیک میتوزی است و ربطی به سن مادر ندارد

-
سندروم ترنر (Turner syndrome)
سندروم ترنر یا مونوزومی کروموزوم X رایج ترین بیماری ژنتیکی در دختران می باشد. این بیماری به دلیل فقدان یک کروموزوم X در دختران ایجاد می شود و آرایش کاریوتایپ افراد مبتلا به صورت 46,XO است. این بیماری ژنتیکی اولین بار توسط دکتر هنری ترنر در سال 1938 شناسایی شد. مونوزومی X تنها منوزومی سازگار با حیات انسان است؛ با این حال 98% جنین های مبتلا به طور خود به خودی سقط می شوند. در این بیماری که فقط زنان را تحت تاثیر قرار می دهد یکی از کروموزوم های X به طور کامل یا جزئی از دست می رود. شیوع این سندروم 1 در 2500 تولد زنده می باشد.
علائم
- قد کوتاه
- گردن پره دار
- خط رویش موی پایین
- تورم دست ها و پاها
- مشکلات قلبی و کلیوی
- فشار خون بالا (به علت مشکلات قلبی)
- کاهش شنوایی (به علت عفونت گوش)
- ناباروری و اختلال در عملکرد تخمدان ها

علت بروز
- ایزوکروموزوم Xq(Isochromosome): در این حالت دو نسخه از بازوی بلند کروموزوم وجود دارد که سر به سر به هم متصل هستند
- کروموزوم حلقوی (Ring chromosome): در این حالت قسمتی از انتهای بازوهای کوتاه و بلند کروموزوم X وجود نداشته و کروموزوم ها حلقوی شده اند
- حذف Xp یا Xq(Xp or Xq deletion): در این موارد حذف بخشی از بازوی کوتاه کروموزوم X رخ داده است
- موزائیسم کروموزوم Y
- 45, X به شکل موزائیسم

-
سندروم کلاین فلتر (Klinefelter syndrome)
این سندروم یک اختلال ژنتیکی مخصوص به مردان است که در آن یک کروموزوم X اضافی وجود دارد و آرایش کاریوتایپ فرد مبتلا به صورت 47,XXY است. از دیگر کاریوتایپ های این بیماری می توان به موزائیسم 46, XY/47, XXy و آنیوپلوئیدی هایی مانند 48, XXXY و 49,XXXXY اشاره کرد. این سندروم شایع ترین اختلال کروموزوم های جنسی است و شیوع این اختلال 1 در هر 500-1000 تولد زنده گزارش شده است که با افزایش سن مادر (35 سال به بالا) احتمال وقوع آن افزایش می یابد. این سندروم اغلب تا بزرگسالی تشخیص داده نمی شود و ممکن است زمانی تشخیص داده شود که مرد متوجه ناباروری خود می شود. کلاین فلتر به دلیل خطا در زمان لقاح به وجود می آید. کلاین فلتر بیماری ارثی نیست و از والدین به فرزندان منتقل نمی شود.
علائم
- ضعف عضلات
- مشکلات یادگیری و صحبت
- بلند قد، لاغر، خجالتی و ساکت
- دارای ضریب هوشی طبیعی
- پا های بلند و لگن پهن
- تاخیر در بلوغ
- قدرت بدنی پائین
- بیضه های کوچک و سفت
- کاهش رویش مو در صورت و بدن
- هایپوگنادیسم
- فقدان اسپرم در مایع منی و ناباروری

- سندروم جاکوب (Jacobs Syndrome)
سندروم جاکوب یا سندروم ابرمرد نوعی آنیوپلوئیدی بسیار نادر است که مردان مبتلا به آن یک کروموزوم Y اضافی از پدر خود دریافت می کنند. آرایش کاریوتایپ این افراد به صورت 47,XYY می باشد. شیوع این بیماری 1 در هر 1000 تولد زنده است. سندروم جاکوب به صورت تصادفی ایجاد می شود و ارثی نیست. عامل این بیماری، خطای عدم انفصال کروموزوم ها در تقسیم میوز 2 هنگام تشکیل سلول های جنسی پدر است که اسپرم تشکیل شده دو نسخه کروموزوم Y دریافت می کند. البته حالت نادری هم وجود دارد که این سندروم به دلیل خطا در تقسیم میتوز طی رشد جنین رخ می دهد و حالت موزاییسم (47,XYY یا 46,XY) ایجاد می شود.
علائم
- قد بلند
- ماکروسفالی
- هایپوتونی یا شل بودن عضلات
- وجود مشکلات در یادگیری و صحبت کردن
- IQ این افراد 10 تا 20 واحد کمتر از برادر یا خواهر سالم خود است
- در معرض خطر ابتلا به اختلال نقص توجه و بیش فعالی، افسردگی و اوتیسم هستند
- این مبتلا به سندروم جاکوب توان باروری نیز دارند اما در مواردی مشکل تکامل بیضه، نقص تولید اسپرم یا تستوسترون مشاهده شده است و بعضی از این بیماران عقیم هستند

علت بروز
- اختلال در میوز II پدری
- موزائیسم 46,XY/47,XYY
-
سندروم XXX (Triple X)
- تریزومی کروموزوم X، سندروم Triple x یا سندروم ابرزن با آرایش کاریوتایپ 47,XXX به حضور یک کروموزوم X اضافه در سلول های بدن اشاره دارد. احتمال وقوع این سندروم 1 در هر 1000 دختر متولد شده است؛ البته بسیاری از موارد ابتلا هرگز تشخیص داده نمی شوند. اگرچه افراد مبتلا به تریزومی X معمولا قد بلندتری دارند، اما معمولا این تغییر کرومزومی تغییرات فیزیکی خاصی ایجاد نمی کند. این سندروم فقط زنان را تحت تاثیر قرار می دهد.
علائم
- افزایش ریسک ناتوانی در یادگیری و صحبت کردن
- تاخیر در تکامل مهارت های یادگیری و حرکت کردن
- هیپوتونی و مشکلات حسی و رفتاری
- کاهش 10 الی 20 واحد IQ و رفتارهای مخالفت گرایانه
- بلوغ زودتر یا دیرتر از موعد
- کوچک بودن سر، شکاف پلکی رو به بالا
- فاصله زیاد بین دو چشم
- تاخوردگی و چین در گوشه چشم
- کف پای صاف
- حجم کم ماهیچه ها
- انحنا در انگشتان

علت بروز
- علت وقوع این سندروم بروز خطا در تشکیل اسپرم پدر یا تخمک مادر است. گاهی این سندروم هنگام تکوین جنین رخ می دهد. در واقع این سندروم در اثر خطای تصادفی ژنتیکی ایجاد می شود و یک بیماری ارثی نیست. در بعضی موارد افراد مبتلا حالت موزاییسم داشته و در بدن آن ها ترکیبی از سلول های 47,XXX و 46,XX وجود دارد.
روش های تشخیص آنیوپلوئیدی در افراد
امروزه با پیشرفت های تکنولوژی، روش های زیادی برای تشخیص ناهنجاری های کروموزومی در دوران جنینی وجود دارد. این روش ها شامل آزمایش های غربالگری و روش های تشخیصی دقیق است. آزمایش های غربالگری شامل سونوگرافی N.T در هفته 11 تا 14 بارداری، آزمایش های سهگانه (اندازه گیری فاکتور های خون مادر) در هفته 15 تا 18 بارداری و انجام آزمایش های ژنتیکی با استفاده از DNA جنینی موجود در سرم خون مادر (NIPT) هستند. آزمایش های غربالگری می توانند در مراحل اولیه کاربردی باشند اما از آنجایی که نتایج آن ها ممکن است دقیق و قطعی نباشد برای اطمینان باید از روش های تشخیصی دقیق تر استفاده شود که معمولا نیازمند نمونه برداری های داخل رحمی هستند. برای انجام این روش های تشخیصی لازم است نمونه برداری از پرزهای جفتی (CVS) بین هفته 8 تا 12 بارداری و یا نمونه برداری از مایع آمنیوتیک (آمنیوسنتز) بین هفته 15 تا 20 بارداری انجام شود. با تهیه کاریوتایپ و یا انجام تکنیک های مولکولی مانند QF-PCR، MLPA و FISH با استفاده از DNA استخراج شده از نمونه های CVS و یا مایع آمنیوتیک می توان وجود یا عدم وجود نا هنجاری های ژنتیکی در جنین را پیش از تولد تشخیص داد.
-
کاریوتایپ
کاریوتایپ تصویری از مجموعه کروموزوم های یک موجود زنده است. برای تهیه این تصویر کروموزوم ها با رنگ های خاصی مانند گیمسا رنگ آمیزی می شوند. طی این رنگ آمیزی هر کروموزوم با الگوی خاصی از نوارهای تیره و روشن موسوم به G-banding رنگ گرفته و مجموعه کروموزوم ها با استفاده از میکرسکوپ قابل رویت می شوند. معمولا نواحی غنی از GC که حاوی ژن ها هستند رنگ تیره به خود می گیرند. با استفاده از این روش بررسی شکل، اندازه و تعداد کروموزوم ها امکان پذیر است.
کاریوتایپینگ کروموزوم انسانی به معنی آنالیز 46 کروموزوم انسان و باندهای G در آن ها، به منظور تشخیص از دست دادن یا به دست آوردن کروموزوم ها، حذف های بزرگ (Deletion)، بازآرایی ها (Translocation & Inversion) و مضاعف شدگی ها (Duplication) است. این بررسی در فشرده ترین حالت کروموزوم یعنی مرحله متافاز تقسیم میتوز انجام می شود؛ بنابراین برای انجام آن، سلول های نمونه مورد نظر با روش کشت سلولی تکثیر می شوند و در مرحله متافاز جمع آوری می گردند. برای این منظور نمونه های مورد نظر با تکنیک کشت سلولی طی چند روز تکثیر می شوند. سپس با اضافه شدن ماده colcemid (colchicine) تقسیم سلولی متوقف می شود و سلول ها در مرحله متافاز بلاک می شوند. کروموزوم های متافازی جدا سازی، رنگ آمیزی و از طریق باند های موجود با استفاده از میکروسکوپ آنالیز می شوند. از این روش برای تشخیص سندروم های مرتبط با آنیوپلوئیدی (سندروم های داون، ادوارد، پاتو، ترنر و …) استفاده می شود. در واقع تهیه کاریوتایپ به عنوان استاندارد طلایی تشخیص ناهنجاری های کروموزومی و تعیین وضعیت آنیوپلوئیدی می باشد.
در صورت مشاهده حذف یا اضافه شدن کروموزوم در کاریوتایپ فرزند، بررسی کاریوتایپ پدر و مادر از نظر وجود ترنسلوکیشن های متعادل (که به فرزندان منتقل می شوند) توصیه می شود. برای تهیه کاریوتایپ می توان از بافت های مختلفی استفاده کرد، اما برای هر بافت محدودیت تشخیص وجود دارد؛ به عنوان مثال برای تشخیص تریزومی یا تترازومی نمی توان از نمونه خون استفاده کرد و نمونه پوست لازم است. به طور معمول برای تشخیص پیش از تولد آنیوپلوئیدی ها از نمونه مایع آمنیوتیک یا پرزهای کویونی استفاده می شود.
با استفاده از کاریوتایپ می توان حذف و اضافه شدن های تا 3-4 ملیون جفت باز را تشخیص داد. تشخیص تغییرات کوچک تر نیازمند روش های سیتوژنیک مولکولی است. تهیه کاریوتایپ به دلیل پیش نیاز کشت سلولی نیازمند زمان معمولا حدود 2 هفته است. اما امروزه با استفاده از تکنیک های مولکولی امکان تشخیص سریع در کمتر از 48 ساعت فراهم شده است. میزان خطای این روش 0.1 تا 0.6 درصد است که بیشتر مربوط به خطای تعیین جنسیت و آن هم به دلیل آلودگی به نمونه مادر است. زمان بر بودن، سختی کار و هزینه بر بودن این روش تمایل به استفاده از سایر روش ها را افزایش داده است. با این حال این روش هنوز به عنوان استاندارد طلایی تشخیص نا هنجاری های کروموزومی در آزمایشگاه های تشخیصی مورد استفاده قرار می گیرد.
-
Quantitative fluorescent PCR
Quantitative fluorescent PCR یا QF-PCR یکی از تکنیک های مولکولی است که از سال 1993 برای تشخیص پیش از تولد آنیوپلوئیدی های کروموزوم های 13، 18، 21، X و Y استفاده شده است. این تکنیک از تنوع توالی های کوتاه تکراری موسوم به STR یا میکروستلایت ها برای شناسایی همولوگ های کروموزومی استفاده می کند. STR ها از نظر تعداد تکرارها و طول توالی روی لوکوس های متفاوت با هم فرق می کنند. شناسایی و کمی سازی STR ها امکان تعیین تعداد کروموزوم ها را فراهم می کند. در این تکنیک پس از استخراج DNA از نمونه (مایع آمنیوتیک یا پرزهای کوریونی)، تکثیر مارکرهای STR روی کروموزوم های مورد نظر (معمولا کروموزوم های 13، 18، 21، X و Y) با استفاده از روش PCR و در دستگاه ترمال سایکلر انجام می شود. پرایمر های مورد استفاده از در این واکنش با مولکول های فلورسنت با رنگ های متفاوت برچسب گذاری شده اند. در نتیجه پس از تکثیر مارکر ها امکان تفکیک محصولات PCR با روش کپیلاری الکتروفورز در دستگاه ژنتیک آنالایزر وجود دارد.
با تفسیر نتایج حاصل از QF-PCR و کپیلاری الکتروفورز امکان بررسی تعداد هر یک از کروموزوم های مورد بررسی فراهم شده است. نتایج حاصل از کپیلاری الکتروفورز با استفاده از نرم افزارهایی مانند Gene mapper و Gene marker به صورت سیگنال هایی قابل مشاهده می شوند که به آن الکتروفوروگرام می گویند. در افراد نرمال که از هر کروموزوم اتوزوم دو نسخه وجود دارد، به ازای مارکر های هتروزیگوت اتوزومال، دو سیگنال با سطح زیر منحنی مساوی ایجاد می گردد. در صورتی که فرد به تریزومی مبتلا باشد، سه نسخه از کروموزوم مربوطه وجود دارد و دو سیگنال با نسبت های 1:2 ، 2:1 و یا سه سیگنال 1:1:1 ایجاد می شود. بنابراین امکان تشخیص آنیوپلوئیدی ها فراهم می شود.
دقت و اختصاصیت این روش بسیار بالاست و در مدت زمان 48 ساعت قادر به جوابدهی است. اما باید به این موضوع توجه داشت که با روش QF-PCR امکان تشخیص بازآرایی های نامتعادل و حالت های موزاییسم آنیوپلوئیدی ها قابل تشخیص نیست؛ از این رو جهت حصول نتایج دقیق تر بهتر است این تکنیک مولکولی در کنار روش سیتوژنتیک (کاریوتایپینگ) به صورت توامان انجام گیرند.
-
MLPA
روش MLPA (Multiplex Ligation-dependent probe amplification) یک روش تشخیص مولکولی حساس و مؤثر برای تشخیص تعداد نسخه های ژنی (حذف و اضافه) ، جهش های نقطه ای و ناهنجاری های کروموزومی می باشد. این روش، نوع تغییر یافته ای از روش Multiplex PCR می باشد که طی آن حداکثر 50 ناحیه ژنی فقط با استفاده از یک جفت پرایمر و با استفاده از پروب های مخصوص تکثیر می گردد و امکان تشخیص و تمایز دادن توالی هایی که تنها در یک نوکلئوتید تفاوت دارند را فراهم می کند.
پروب های MLPA به صورت دو نیمه پروب هستند که حاوی قطعات مکمل با DNA، مکمل با پرایمر و Stuffer می باشند. تفاوت طول توالی Stuffer امکان جداسازی قطعات تکثیر شده را فراهم می کند. این تکنیک در تشخیص سرطان ها، جهش ها، ناقلین بیماری های ژنتیک به خصوص تالاسمی آلفا و بتا و دستروفی عضلانی دوشن و … کاربرد دارد. در سال های اخیر، استفاده از MLPA برای تشخیص آنیوپلوئیدی کروموزوم های 13، 18، 21، X و Y با استفاده از نمونه های مایع آمنیوتیک و پرزهای کوریونی پیشنهاد شده است.
انجام این تکنیک شامل 5 مرحله است؛ ابتدا DNA واسرشته می شود و پروب ها (به صورت دو نیمه پروب) با توالی مکمل خود روی DNA هیبرید می شوند. در مرحله بعد، در صورت صحیح بودن توالی و عدم وجود جهش ها، دو نیمه پروب با آنزیم لیگاز به یکدیگر متصل (ligation) می شوند. سپس پروب های ligation شده با استفاده از پرایمرهای نشاندار تکثیر می شوند و در نهایت محصولات تکثیر شده با روش کپیلاری الکتروفورز تفکیک شده و نتایج آنالیز می شوند. مقایسه ارتفاع و سطح زیر پیک های حاصل از کپیلاری الکتروفورز نمونه مورد بررسی با کنترل های استاندارد، طبیعی یا غیر طبیعی بودن تعداد نسخه های مورد بررسی را آشکار می سازد. از مزایای این روش می توان به دقت و اختصاصیت بالا، نیاز به میزان کم DNA، هزینه مناسب و نیاز به تنها یک جفت پرایمر اشاره کرد. با استفاده از این تکنیک می توان جهش های کوچک که روش FISH قادر به تشخیص آن نیست را شناسایی کرد. توان عملیاتی این روش بالاست و نتایج آن در مدت 24 ساعت آماده می شود. با این حال این روش قادر به تشخیص سلول های پلی پلوئید و ترنسلوکاسیون های متعادل نمی باشد.
-
NGS
در دهه های اخیر با ظهور تکنیک Next Generation Sequencing یا NGS توالی یابی ژن ها و تشخیص بیماری های ژنتیکی وارد عرصه های جدیدی شده است. پیشرفت هایی که امروزه در زمینه توالی یابی DNA رخ داده است، باعث به وجود آمدن تکنیک جدیدی به نام توالی یابی نسل جدید یا NGS شده است. در این تکنیک که مستقل از سلول است و نیازی به اقدامات پس از تکثیر یعنی الکتروفورز ندارد، ابتدا DNA ژنومی به قطعات کوچک تبدیل می شوند و کتابخوانه ای از قطعات DNA حاصل می شود. پس از آن الیگونوکلئوتیدهایی با توالی مشخص به انتهای این قطعات متصل می گردند و قطعات تکثیر می شوند.
در این روش میلیون ها واکنش توالی یابی به طور هم زمان و به موازات هم انجام می شوند. این تکنیک در شناسایی آنیوپلوئیدی پس از نمونه برداری از بلاستوسیت استفاده می شود. برای آنالیز پیش از تولد تعداد کپی کروموزوم ها با استفاده از NGS، باید DNA رویانی به قطعات کوچک 100-200 bp تبدیل شود. صدها هزار از این قطعات کوچک به طور موازی توالی یابی می شوند. نتایج این توالی یابی ها ابتدا با ژنوم مرجع مقایسه می شود و سپس با نرم افزارهای مخصوص محاسبه می گردد. از آنجایی که عدد توالی های یک کروموزوم باید متناسب با تعداد کپی باشد، تریزومی یا منوزومی به ترتیب منجر به افزایش و کاهش عددهای خوانش ها می شود.
با استفاده از این روش و نمونه های بیوپسی بلاستومر، ناهنجاری های ساختاری و آنیوپلوئیدی قابل تشخیص است. این روش در مطالعات مختلف با سایر روش های تشخیص آنیوپلوئیدی صحه گذاری شده است و نشان دهنده دقت صد در صدی است. این روش نه تنها آنیوپلوئیدی را تشخیص میدهد، بلکه اختلالات تک ژنی، ترنسلوکیشن ها و اختلالات ژنوم میتوکندری را شناسایی می کند. با این حال از معایب NGS این است که امکان تشخیص ناهجاری های ساختاری نامتعادل را ندارد.
درمان آنیوپلوئیدی ها
به طور کلی، بیماری های آنیوپلوئیدی مانند سندرم داون مادر زادی بوده و نوزاد در زمان تولد به آن مبتلا می باشد. اغلب آنیوپلوئیدی ها برای نوزاد کشنده بوده و یا اینکه منجر به بروز نفص های مادرزادی مانند ناتوانی های ذهنی خواهند شد که قابل درمان نیستند. از سوی دیگر، زنان بارداری که جنین آنها مبتلا به ناهنجاری های کروموزومی است، خطر سقط جنین بالایی دارند. با توجه به اینکه تا امروز روش درمانی موثری برای این بیماری ها ارائه نشده است، تنها می توان به رعایت برخی مسائل مانند داشتن رژیم غذایی سالم، عدم استعمال دخانیات و انجام منظم آزمایشات غربالگری در طول دوران بارداری خطر تولد نوزاد مبتلا به این دسته از ناهنجاری های ژنتیکی را کاهش داد.
منابع علمی
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4714037
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2997176
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3613689
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3551553/table/T1/?report=objectonly
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3551553
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2429870
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3293208
https://www.ncbi.nlm.nih.gov/books/NBK570597
https://www.ncbi.nlm.nih.gov/books/NBK538347
https://www.ncbi.nlm.nih.gov/books/NBK554621
https://www.ncbi.nlm.nih.gov/books/NBK482314
https://www.ncbi.nlm.nih.gov/books/NBK557699
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3632021/pdf/NAJMS-5-182.pdf
